1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
470
471
472
473
474
475
476
477
478
479
480
481
482
483
484
485
486
487
488
489
490
491
492
493
494
495
496
497
498
499
500
501
502
503
504
505
506
507
508
509
510
511
512
513
514
515
516
517
518
519
520
521
522
523
524
525
526
527
528
529
530
531
532
533
534
535
536
537
538
539
540
541
542
543
544
545
546
547
548
549
550
551
552
553
554
555
556
557
558
559
560
561
562
563
564
565
566
567
568
569
570
571
572
573
574
575
576
577
578
579
580
581
582
583
584
585
586
587
588
589
590
591
592
593
594
595
596
597
598
599
600
601
602
603
604
605
606
607
608
609
610
611
612
613
614
615
616
617
618
619
620
621
622
623
624
625
626
627
628
629
630
631
632
633
634
635
636
637
638
639
640
641
642
643
644
645
646
647
648
649
650
651
652
653
654
655
656
657
658
659
660
661
662
663
664
665
666
667
668
669
670
671
672
673
674
675
676
677
678
679
680
681
682
683
684
685
686
687
688
689
690
691
692
693
694
695
696
697
698
699
700
701
702
703
704
705
706
707
708
709
710
711
712
713
714
715
716
|
% bedtools Tutorial 2017-2018, Hao Hou
% Aaron Quinlan
Synopsis
========
Our goal is to work through examples that demonstrate how to
explore, process and manipulate genomic interval files (e.g., BED, VCF, BAM) with the `bedtools` software package.
Some of our analysis will be based upon the Maurano et al exploration of DnaseI hypersensitivity sites in hundreds of primary tissue types.
Maurano et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science. 2012. Vol. 337 no. 6099 pp. 1190-1195.
www.sciencemag.org/content/337/6099/1190.short
This tutorial is merely meant as an introduction to whet your appetite. There are many, many more tools and options than presented here. We therefore encourage you to read the bedtools [documentation](http://bedtools.readthedocs.org/en/latest/).
<div class="alert alert-info" role="alert">NOTE: We recommend making your browser window as large as possible because some of the examples yield "wide" results and more screen real estate will help make the results clearer.-</div>
\
Setup
=====
From the Terminal, create a new directory on your Desktop called `bedtools-demo` (it doesn't really matter where you create this directory).
cd ~/Desktop
mkdir bedtools-demo
Navigate into that directory.
cd bedtools-demo
Download the sample BED files I have provided.
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/maurano.dnaseI.tgz
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/cpg.bed
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/exons.bed
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/gwas.bed
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/genome.txt
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/hesc.chromHmm.bed
Now, we need to extract all of the 20 Dnase I hypersensitivity BED files from the "tarball" named
`maurano.dnaseI.tgz`.
tar -zxvf maurano.dnaseI.tgz
rm maurano.dnaseI.tgz
Let's take a look at what files we now have.
ls -1
\
What are these files?
=========================
Your directory should now contain 23 BED files and 1 genome file. Twenty of these files (those starting with "f" for "fetal tissue") reflect Dnase I hypersensitivity sites measured in twenty different fetal tissue samples from the brain, heart, intestine, kidney, lung, muscle, skin, and stomach.
In addition: `cpg.bed` represents CpG islands in the human genome; `exons.bed` represents RefSeq exons from human genes; `gwas.bed` represents human disease-associated SNPs that were identified in genome-wide association studies (GWAS); `hesc.chromHmm.bed` represents the predicted function (by chromHMM) of each interval in the genome of a human embryonic stem cell based upon ChIP-seq experiments from ENCODE.
The latter 4 files were extracted from the UCSC Genome Browser's [Table Browser](http://genome.ucsc.edu/cgi-bin/hgTables?command=start).
In order to have a rough sense of the data, let's load the `cpg.bed`, `exons.bed`, `gwas.bed`, and `hesc.chromHmm.bed` files into [IGV](http://www.broadinstitute.org/igv/). To do this, launch IGV, then click File->Load from File. Then select the four files. IGV will warn you that you need to create an index for a couple of the files. Just click OK, as these indices are created automatically and speed up the processing for IGV.
Once loaded, navigate to `TP53` by typing `TP53` in the search bar. Change the track height to 200 for each track, set the font size to 16 for each track, and change the track colors to match the following image:

Visualization in IGV or other browsers such as UCSC is a tremendously useful way to make sure that your results make sense to your eye. Coveniently, a subset of bedtools is built-into IGV!
\
The bedtools help
==================
Bedtools is a command-line tool. To bring up the help, just type
bedtools
As you can see, there are multiple "subcommands" and for bedtools to
work you must tell it which subcommand you want to use. Examples:
bedtools intersect
bedtools merge
bedtools subtract
What version am I using?
bedtools --version
How can I get more help?
bedtools --contact
bedtools "intersect"
====================
The `intersect` command is the workhorse of the `bedtools` suite. It compares two or more BED/BAM/VCF/GFF files and identifies all the regions in the genome where the features in the two files overlap (that is, share at least one base pair in common).
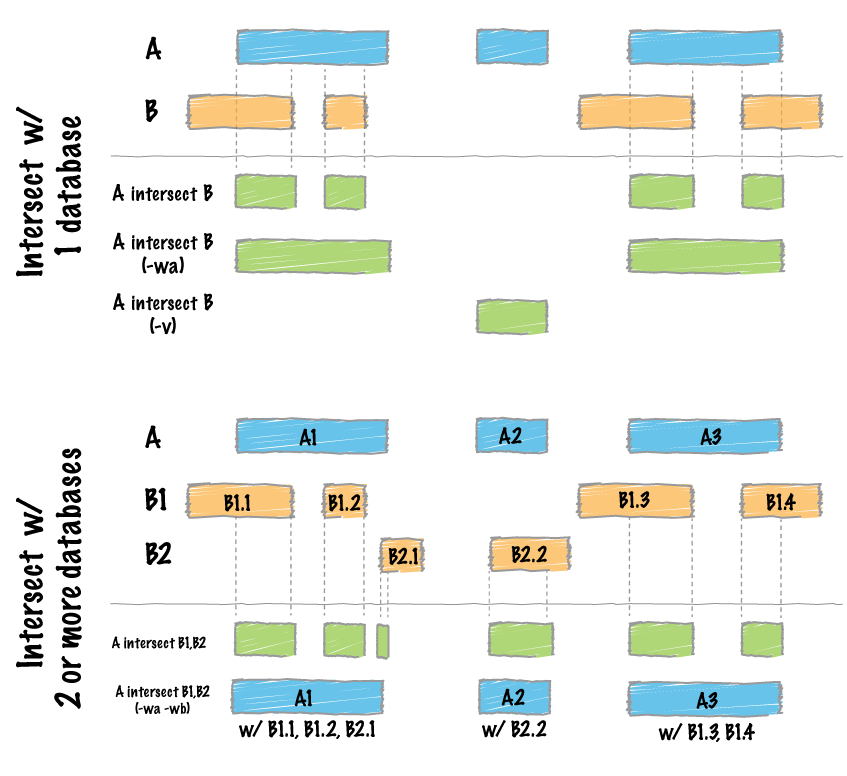
Default behavior
----------------
By default, `intersect` reports the intervals that represent overlaps between your two files. To demonstrate, let's identify all of the CpG islands that overlap exons.
bedtools intersect -a cpg.bed -b exons.bed | head -5
chr1 29320 29370 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
<div class="alert alert-info" role="alert">NOTE: In this case, the intervals reported are NOT the original CpG intervals, but rather a refined interval reflecting solely the portion of each original CpG interval that overlapped with the exon(s).</div>
Reporting the original feature in each file.
--------------------------------------------
The `-wa` (write A) and `-wb` (write B) options allow one to see the original records from the A and B files that overlapped. As such, instead of not only showing you *where* the intersections occurred, it shows you *what* intersected.
bedtools intersect -a cpg.bed -b exons.bed -wa -wb \
| head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r -
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 -
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 +
How many base pairs of overlap were there?
------------------------------------------
The `-wo` (write overlap) option allows one to also report the *number* of base pairs of overlap between the features that overlap between each of the files.
bedtools intersect -a cpg.bed -b exons.bed -wo \
| head -10
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r - 50
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 439
chr1 713984 714547 CpG:_60 chr1 713663 714068 NR_033908_exon_6_0_chr1_713664_r 0 84
chr1 762416 763445 CpG:_115 chr1 761585 762902 NR_024321_exon_0_0_chr1_761586_r - 486
chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_015368_exon_0_0_chr1_762971_f + 185
chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_047519_exon_0_0_chr1_762971_f + 185
chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_047520_exon_0_0_chr1_762971_f + 185
Counting the number of overlapping features.
--------------------------------------------
We can also count, for each feature in the "A" file, the number of overlapping features in the "B" file. This is handled with the `-c` option.
bedtools intersect -a cpg.bed -b exons.bed -c \
| head
chr1 28735 29810 CpG:_116 1
chr1 135124 135563 CpG:_30 1
chr1 327790 328229 CpG:_29 3
chr1 437151 438164 CpG:_84 0
chr1 449273 450544 CpG:_99 0
chr1 533219 534114 CpG:_94 0
chr1 544738 546649 CpG:_171 0
chr1 713984 714547 CpG:_60 1
chr1 762416 763445 CpG:_115 10
chr1 788863 789211 CpG:_28 9
\
Find features that DO NOT overlap
--------------------------------------------
Often we want to identify those features in our A file that **do not** overlap features in the B file. The `-v` option is your friend in this case.
bedtools intersect -a cpg.bed -b exons.bed -v \
| head
chr1 437151 438164 CpG:_84
chr1 449273 450544 CpG:_99
chr1 533219 534114 CpG:_94
chr1 544738 546649 CpG:_171
chr1 801975 802338 CpG:_24
chr1 805198 805628 CpG:_50
chr1 839694 840619 CpG:_83
chr1 844299 845883 CpG:_153
chr1 912869 913153 CpG:_28
chr1 919726 919927 CpG:_15
Require a minimal fraction of overlap.
--------------------------------------------
Recall that the default is to report overlaps between features in A and B so long as *at least one basepair* of overlap exists. However, the `-f` option allows you to specify what fraction of each feature in A should be overlapped by a feature in B before it is reported.
Let's be more strict and require 50% of overlap.
bedtools intersect -a cpg.bed -b exons.bed \
-wo -f 0.50 \
| head
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 439
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047519_exon_5_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047521_exon_4_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047523_exon_3_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047524_exon_3_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047525_exon_4_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788858 794826 NR_047520_exon_6_0_chr1_788859_f 0 348
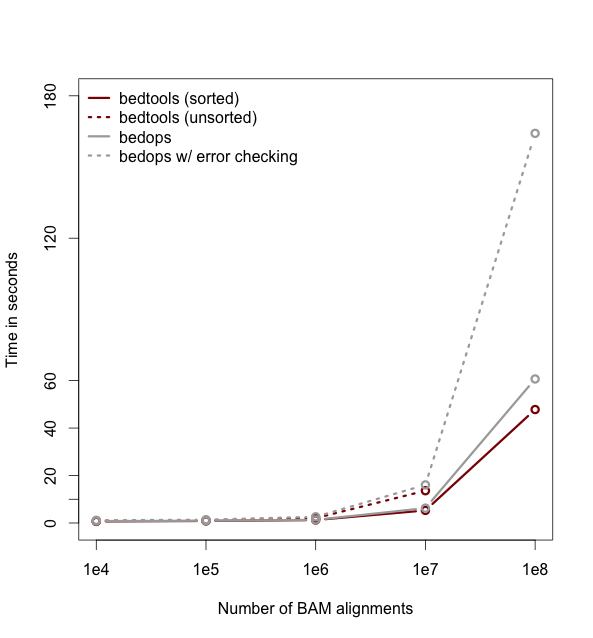
Faster analysis via sorted data.
--------------------------------------------
So far the examples presented have used the traditional algorithm in bedtools for finding intersections. It turns out, however, that bedtools is much faster when using presorted data.
For example, compare the difference in speed between the two approaches when finding intersections
between `exons.bed` and `hesc.chromHmm.bed`:
time bedtools intersect -a gwas.bed -b hesc.chromHmm.bed > /dev/null
1.10s user 0.11s system 99% cpu 1.206 total
time bedtools intersect -a gwas.bed -b hesc.chromHmm.bed -sorted > /dev/null
0.36s user 0.01s system 99% cpu 0.368 total
<div class="alert alert-info" role="alert">NOTE: While the run times in this example are quite small, the performance gains from using the `-sorted` option groqw as datasets grow larger. For example, compare the runtimes of the sorted and unsorted approaches as a function of dataset size in the figure below. The important thing to
remember is that each dataset must be sorted by chromosome and then by start position: `sort -k1,1 -k2,2n`.-</div>
Intersecting multiple files at once.
--------------------------------------------
As of version 2.21.0, bedtools is able to intersect an "A" file against one or more "B" files. This greatly
simplifies analyses involving multiple datasets relevant to a given experiment. For example, let's intersect
exons with CpG islands, GWAS SNPs, an the ChromHMM annotations.
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted | head
chr1 11873 11937 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 11937 12137 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12137 12227 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 +
chr1 13220 14137 NR_046018_exon_2_0_chr1_13221_f 0 +
chr1 14137 14409 NR_046018_exon_2_0_chr1_13221_f 0 +
chr1 14361 14829 NR_024540_exon_0_0_chr1_14362_r 0 -
chr1 14969 15038 NR_024540_exon_1_0_chr1_14970_r 0 -
chr1 15795 15947 NR_024540_exon_2_0_chr1_15796_r 0 -
chr1 16606 16765 NR_024540_exon_3_0_chr1_16607_r 0 -
Now by default, this isn't incredibly informative as we can't tell which of the three "B" files yielded the intersection with each exon. However, if we use the `-wa` and `wb` options, we can see from which file number (following the order of the files given on the command line) the intersection came. In this case, the 7th column reflects this file number.
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb \
| head -10000 \
| tail -10
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + 3 chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + 3 chr1 27635013 27635413 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27632613 27632813 6_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27632813 27633213 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27635013 27635413 7_Weak_Enhancer
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + 1 chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + 3 chr1 27648613 27649413 1_Active_Promoter
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + 1 chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + 3 chr1 27648613 27649413 1_Active_Promoter
Additionally, one can use file "labels" instead of file numbers to facilitate interpretation, especially when there are _many_ files involved.
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb -names cpg gwas chromhmm \
| head -10000 \
| tail -10
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27635013 27635413 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27632613 27632813 6_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27632813 27633213 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27635013 27635413 7_Weak_Enhancer
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + cpg chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + chromhmm chr1 27648613 27649413 1_Active_Promoter
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + cpg chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + chromhmm chr1 27648613 27649413 1_Active_Promoter
\
bedtools "merge"
====================
Many datasets of genomic features have many individual features that overlap one another (e.g. aligments from a ChiP seq experiment). It is often useful to just cobine the overlapping into a single, contiguous interval. The bedtools `merge` command will do this for you.
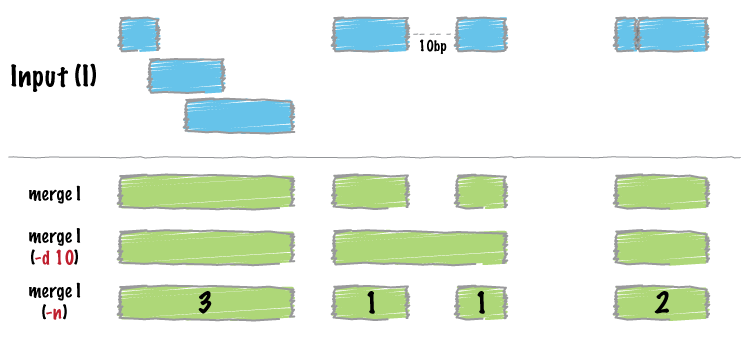
Input must be sorted
--------------------
The merge tool requires that the input file is sorted by chromosome, then by start position. This allows the merging algorithm to work very quickly without requiring any RAM.
If your files are unsorted, the `merge` tool will raise an error. To correct this, you need to sort your BED using the UNIX `sort` utility. For example:
sort -k1,1 -k2,2n foo.bed > foo.sort.bed
Merge intervals.
----------------
Merging results in a new set of intervals representing the merged set of intervals in the input. That is, if a base pair in the genome is covered by 10 features, it will now only be represented once in the output file.
bedtools merge -i exons.bed | head -n 20
chr1 11873 12227
chr1 12612 12721
chr1 13220 14829
chr1 14969 15038
chr1 15795 15947
chr1 16606 16765
chr1 16857 17055
chr1 17232 17368
chr1 17605 17742
chr1 17914 18061
chr1 18267 18366
chr1 24737 24891
chr1 29320 29370
chr1 34610 35174
chr1 35276 35481
chr1 35720 36081
chr1 69090 70008
chr1 134772 139696
chr1 139789 139847
chr1 140074 140566
Count the number of overlapping intervals.
------------------------------------------
A more sophisticated approach would be to not only merge overlapping intervals, but also report the *number* of intervals that were integrated into the new, merged interval. One does this with the `-c` and `-o` options. The `-c` option allows one to specify a column or columns in the input that you wish to summarize. The `-o` option defines the operation(s) that you wish to apply to each column listed for the `-c` option. For example, to count the number of overlapping intervals that led to each of the new "merged" intervals, one
will "count" the first column (though the second, third, fourth, etc. would work just fine as well).
bedtools merge -i exons.bed -c 1 -o count | head -n 20
chr1 11873 12227 1
chr1 12612 12721 1
chr1 13220 14829 2
chr1 14969 15038 1
chr1 15795 15947 1
chr1 16606 16765 1
chr1 16857 17055 1
chr1 17232 17368 1
chr1 17605 17742 1
chr1 17914 18061 1
chr1 18267 18366 1
chr1 24737 24891 1
chr1 29320 29370 1
chr1 34610 35174 2
chr1 35276 35481 2
chr1 35720 36081 2
chr1 69090 70008 1
chr1 134772 139696 1
chr1 139789 139847 1
chr1 140074 140566 1
Merging features that are close to one another.
-----------------------------------------------
With the `-d` (distance) option, one can also merge intervals that do not overlap, yet are close to one another. For example, to merge features that are no more than 1000bp apart, one would run:
bedtools merge -i exons.bed -d 1000 -c 1 -o count | head -20
chr1 11873 18366 12
chr1 24737 24891 1
chr1 29320 29370 1
chr1 34610 36081 6
chr1 69090 70008 1
chr1 134772 140566 3
chr1 323891 328581 10
chr1 367658 368597 3
chr1 621095 622034 3
chr1 661138 665731 3
chr1 700244 700627 1
chr1 701708 701767 1
chr1 703927 705092 2
chr1 708355 708487 1
chr1 709550 709660 1
chr1 713663 714068 1
chr1 752750 755214 2
chr1 761585 763229 10
chr1 764382 764484 9
chr1 776579 778984 1
Listing the name of each of the exons that were merged.
-------------------------------------------------------
Many times you want to keep track of the details of exactly which intervals were merged. One way to do this is to create a list of the names of each feature. We can do with with the `collapse` operation available via the `-o` argument. The name of the exon is in the fourth column, so we ask `merge` to create a list of the exon names with `-c 4 -o collapse`:
bedtools merge -i exons.bed -d 90 -c 1,4 -o count,collapse | head -20
chr1 11873 12227 1 NR_046018_exon_0_0_chr1_11874_f
chr1 12612 12721 1 NR_046018_exon_1_0_chr1_12613_f
chr1 13220 14829 2 NR_046018_exon_2_0_chr1_13221_f,NR_024540_exon_0_0_chr1_14362_r
chr1 14969 15038 1 NR_024540_exon_1_0_chr1_14970_r
chr1 15795 15947 1 NR_024540_exon_2_0_chr1_15796_r
chr1 16606 16765 1 NR_024540_exon_3_0_chr1_16607_r
chr1 16857 17055 1 NR_024540_exon_4_0_chr1_16858_r
chr1 17232 17368 1 NR_024540_exon_5_0_chr1_17233_r
chr1 17605 17742 1 NR_024540_exon_6_0_chr1_17606_r
chr1 17914 18061 1 NR_024540_exon_7_0_chr1_17915_r
chr1 18267 18366 1 NR_024540_exon_8_0_chr1_18268_r
chr1 24737 24891 1 NR_024540_exon_9_0_chr1_24738_r
chr1 29320 29370 1 NR_024540_exon_10_0_chr1_29321_r
chr1 34610 35174 2 NR_026818_exon_0_0_chr1_34611_r,NR_026820_exon_0_0_chr1_34611_r
chr1 35276 35481 2 NR_026818_exon_1_0_chr1_35277_r,NR_026820_exon_1_0_chr1_35277_r
chr1 35720 36081 2 NR_026818_exon_2_0_chr1_35721_r,NR_026820_exon_2_0_chr1_35721_r
chr1 69090 70008 1 NM_001005484_exon_0_0_chr1_69091_f
chr1 134772 139696 1 NR_039983_exon_0_0_chr1_134773_r
chr1 139789 139847 1 NR_039983_exon_1_0_chr1_139790_r
chr1 140074 140566 1 NR_039983_exon_2_0_chr1_140075_r
\
bedtools "complement"
=====================
We often want to know which intervals of the genome are **NOT** "covered" by intervals in a given feature file. For example, if you have a set of ChIP-seq peaks, you may also want to know which regions of the genome are not bound by the factor you assayed. The `complement` addresses this task.
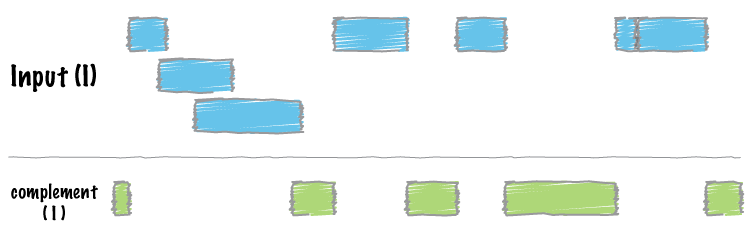
As an example, let's find all of the non-exonic (i.e., intronic or intergenic) regions of the genome. Note, to do this you need a ["genome"](http://bedtools.readthedocs.org/en/latest/content/general-usage.html#genome-file-format) file, which tells `bedtools` the length of each chromosome in your file. *Consider why the tool would need this information...*
bedtools complement -i exons.bed -g genome.txt \
> non-exonic.bed
head non-exonic.bed
chr1 0 11873
chr1 12227 12612
chr1 12721 13220
chr1 14829 14969
chr1 15038 15795
chr1 15947 16606
chr1 16765 16857
chr1 17055 17232
chr1 17368 17605
chr1 17742 17914
\
bedtools "genomecov"
====================
For many analyses, one wants to measure the genome wide coverage of a feature file. For example, we often want to know what fraction of the genome is covered by 1 feature, 2 features, 3 features, etc. This is frequently crucial when assessing the "uniformity" of coverage from whole-genome sequencing. This is done with the versatile `genomecov` tool.
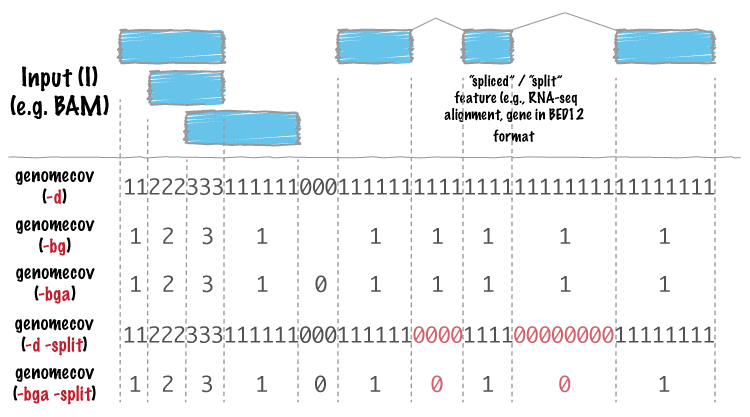
As an example, let's produce a histogram of coverage of the exons throughout the genome. Like the `merge` tool, `genomecov` requires pre-sorted data. It also needs a genome file as above.
bedtools genomecov -i exons.bed -g genome.txt
This should run for 3 minutes or so. At the end of your output, you should see something like:
genome 0 3062406951 3137161264 0.976171
genome 1 44120515 3137161264 0.0140638
genome 2 15076446 3137161264 0.00480576
genome 3 7294047 3137161264 0.00232505
genome 4 3650324 3137161264 0.00116358
genome 5 1926397 3137161264 0.000614057
genome 6 1182623 3137161264 0.000376972
genome 7 574102 3137161264 0.000183
genome 8 353352 3137161264 0.000112634
genome 9 152653 3137161264 4.86596e-05
genome 10 113362 3137161264 3.61352e-05
genome 11 57361 3137161264 1.82844e-05
genome 12 52000 3137161264 1.65755e-05
genome 13 55368 3137161264 1.76491e-05
genome 14 19218 3137161264 6.12592e-06
genome 15 19369 3137161264 6.17405e-06
genome 16 26651 3137161264 8.49526e-06
genome 17 9942 3137161264 3.16911e-06
genome 18 13442 3137161264 4.28477e-06
genome 19 1030 3137161264 3.28322e-07
genome 20 6329 3137161264 2.01743e-06
...
\
Producing BEDGRAPH output
--------------------------
Using the `-bg` option, one can also produce BEDGRAPH output which represents the "depth" fo feature coverage for each base pair in the genome:
bedtools genomecov -i exons.bed -g genome.txt -bg | head -20
chr1 11873 12227 1
chr1 12612 12721 1
chr1 13220 14361 1
chr1 14361 14409 2
chr1 14409 14829 1
chr1 14969 15038 1
chr1 15795 15947 1
chr1 16606 16765 1
chr1 16857 17055 1
chr1 17232 17368 1
chr1 17605 17742 1
chr1 17914 18061 1
chr1 18267 18366 1
chr1 24737 24891 1
chr1 29320 29370 1
chr1 34610 35174 2
chr1 35276 35481 2
chr1 35720 36081 2
chr1 69090 70008 1
chr1 134772 139696 1
\
Sophistication through chaining multiple bedtools
=================================================
Analytical power in `bedtools` comes from the ability to "chain" together multiple tools in order to construct rather sophisicated analyses with very little programming - you just need **genome arithmetic**! Have a look at the examples [here](http://bedtools.readthedocs.org/en/latest/content/advanced-usage.html).
Here are a few more examples.
1. Identify the portions of intended capture intervals that did not have any coverage:
<blockquote class="twitter-tweet" lang="en"><p><a href="https://twitter.com/brent_p">@brent_p</a> bedtools genomecov -ibam aln.bam -bga \ | awk '$4==0' | | bedtools intersect -a regions -b - > foo</p>— Aaron Quinlan (@aaronquinlan) <a href="https://twitter.com/aaronquinlan/status/421786507511205888">January 10, 2014</a></blockquote>
<script async src="//platform.twitter.com/widgets.js" charset="utf-8"></script>
2. [Assessing the breadth and depth coverage of sequencing coverage in exome studies](http://gettinggeneticsdone.blogspot.com/2014/03/visualize-coverage-exome-targeted-ngs-bedtools.html).
\
Principal component analysis
=============================
We will use the bedtools implementation of a Jaccard statistic to meaure the similarity of two
datasets. Briefly, the Jaccard statistic measures the ratio of the number of *intersecting* base
pairs to the *total* number of base pairs in the two sets. As such, the score ranges from 0.0 to 1.
0; lower values reflect lower similarity, whereas higher values reflect higher similarity.
Let's walk through an example: we would expect the Dnase hypersensivity sites to be rather similar
between two samples of the **same** fetal tissue type. Let's test:
bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fHeart-DS15839.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
81269248 160493950 0.50637 130852
But what about the similarity of two **different** tissue types?
bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fSkin_fibro_bicep_R-DS19745.hg19.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
28076951 164197278 0.170995 73261
Hopefully this demonstrates how the Jaccard statistic can be used as a simple statistic to reduce
the dimensionality of the comparison between two large (e.g., often containing thousands or
millions of intervals) feature sets.
\
A Jaccard statistic for all 400 pairwise comparisons.
------------------------------------------------------
We are going to take this a bit further and use the Jaccard statistic to measure the similarity of
all 20 tissue samples against all other 20 samples. Once we have a 20x20 matrix of similarities,
we can use dimensionality reduction techniques such as hierarchical clustering or principal
component analysis to detect higher order similarities among **all** of the datasets.
We will use GNU parallel to compute a Jaccard statistic for the 400 (20*20) pairwise comparisons
among the fetal tissue samples.
But first, we need to install [GNU parallel](http://www.gnu.org/software/parallel/).
brew install parallel
Next, we need to install a tiny script I wrote for this analysis.
curl -O http://quinlanlab.cs.virginia.edu/cshl2013/make-matrix.py
Now, we can use `parallel` to, you guessed it, compute the 400 pairwise Jaccard statistics in parallel using as many processors as you have available.
parallel "bedtools jaccard -a {1} -b {2} \
| awk 'NR>1' \
| cut -f 3 \
> {1}.{2}.jaccard" \
::: *.merge.bed ::: *.merge.bed
This command will create a single file containing the pairwise Jaccard measurements from all 400 tests.
find . \
| grep jaccard \
| xargs grep "" \
| sed -e s"/\.\///" \
| perl -pi -e "s/.bed./.bed\t/" \
| perl -pi -e "s/.jaccard:/\t/" \
> pairwise.dnase.txt
A bit of cleanup to use more intelligible names for each of the samples.
cat pairwise.dnase.txt \
| sed -e 's/.hotspot.twopass.fdr0.05.merge.bed//g' \
| sed -e 's/.hg19//g' \
> pairwise.dnase.shortnames.txt
Now let's make a 20x20 matrix of the Jaccard statistic. This will allow the data to play nicely with R.
awk 'NF==3' pairwise.dnase.shortnames.txt \
| awk '$1 ~ /^f/ && $2 ~ /^f/' \
| python3 make-matrix.py \
> dnase.shortnames.distance.matrix
Let's also make a file of labels for each dataset so that we can label each dataset in our R plot.
cut -f 1 dnase.shortnames.distance.matrix | cut -f 1 -d "-" | cut -f 1 -d "_" > labels.txt
Now start up R.
R
<div class="alert alert-info" role="alert">NOTE: The following example assumes that you have both the `ggplot2` and `RColorBrewer` packages installed on your computer. If they
are not installed, run both `install.packages("ggplot2")` and `install.packages("RColorBrewer")` from the R prompt and respond to the prompts that will follow.-</div>
You should see something very similar to this:
R version 2.15.1 (2012-06-22) -- "Roasted Marshmallows"
Copyright (C) 2012 The R Foundation for Statistical Computing
ISBN 3-900051-07-0
Platform: x86_64-apple-darwin12.0.0 (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
Natural language support but running in an English locale
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
>
No paste these commands into the R console:
library(ggplot2)
library(RColorBrewer)
blues <- colorRampPalette(c('dark blue', 'light blue'))
greens <- colorRampPalette(c('dark green', 'light green'))
reds <- colorRampPalette(c('pink', 'dark red'))
setwd("~/Desktop/bedtools-demo")
x <- read.table('dnase.shortnames.distance.matrix')
labels <- read.table('labels.txt')
ngroups <- length(unique(labels))
pca <- princomp(x)
qplot(pca$scores[,1], pca$scores[,2], color=labels[,1], geom="point", size=1) +
scale_color_manual(values = c(blues(4), greens(5), reds(5)))
You should see this:

Et voila.
<div class="alert alert-info" role="alert">Note that PCA was used in this case as a toy example of what PCA does for the CSHL Adv. Seq. course. Heatmaps are a more informative visualization in this case since Jaccard inherently returns a measure of distance.</div>
So let's make a heatmap for giggles.
<div class="alert alert-info" role="alert">NOTE: The following example assumes that you have both the `gplots` package installed on your computer. If it
are not installed, run `install.packages("gplots")` from the R prompt and respond to the prompts that will follow.-</div>
library(gplots)
# thanks for the fix, Stefanie Dukowic-Schulze
jaccard_table <- x
jaccard_matrix <- as.matrix(jaccard_table)
heatmap.2(jaccard_matrix, col = brewer.pal(9,"Blues"), margins = c(14, 14), density.info = "none", lhei=c(2, 8), trace= "none")
You should see this:

\
Puzzles to help teach you more bedtools.
========================================
1. Create a BED file representing all of the intervals in the genome
that are NOT exonic and are not Promoters (based on the promoters in the hESC file).
2. What is the average distance from GWAS SNPs to the closest exon? (Hint - have a look at the [closest](http://bedtools.readthedocs.org/en/latest/content/tools/closest.html) tool.)
3. Count how many exons occur in each 500kb interval ("window") in the human genome. (Hint - have a look at the `makewindows` tool.)
4. Are there any exons that are completely overlapped by an enhancer? If so, how many?
5. What fraction of the GWAS SNPs are exonic? Hint: should you worry about double counting?
6. What fraction of the GWAS SNPs are lie in either enhancers or promoters in the hESC data we have?
7. Create intervals representing the canonical 2bp splice sites on either side of each exon (don't worry about excluding splice sites at the first or last exon). (Hint - have a look at the [flank](http://bedtools.readthedocs.org/en/latest/content/tools/flank.html) tool.)
8. What is the Jaccard statistic between CpG and hESC enhancers? Compare that to the Jaccard statistic between CpG and hESC promoters. Does the result make sense? (Hint - you will need `grep`).
9. What would you expect the Jaccard statistic to look like if promoters were randomly distributed throughout the genome? (Hint - you will need the [shuffle](http://bedtools.readthedocs.org/en/latest/content/tools/shuffle.html) tool.)
10. Which hESC ChromHMM state (e.g., 11_Weak_Txn, 10_Txn_Elongation) represents the most number of base pairs in the genome? (Hint: you will need to use `awk` or `perl` here, as well as the [groupby](http://bedtools.readthedocs.org/en/latest/content/tools/groupby.html) tool.)
[answers](http://quinlanlab.org/tutorials/cshl2014/answers.html)
|
