1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
470
|
# Analyzing the genomic and taxonomic composition of an environmental genome using GTDB and sample-specific MAGs with sourmash
C. Titus Brown, Taylor Reiter, and Tessa Pierce Ward
July 2022
Based on a tutorial developed for MBL STAMPS 2022.
You'll need 5 GB of disk space and 5 GB of RAM in order to run this tutorial.
It will take about 30 minutes of compute time to execute all the commands.
---
```{contents}
:depth: 2
```
In this tutorial, we'll use sourmash to analyze the composition of a metagenome, both genomically and taxonomically. We'll also use sourmash to classify some MAGs and integrate them into our analysis.
## Install sourmash
First, we need to install the software! We'll use conda/mamba to do this.
The below command installs [sourmash](http://sourmash.readthedocs.io/) and [GNU parallel](https://www.gnu.org/software/parallel/).
Run:
```
# create a new environment
mamba create -n smash -y -c conda-forge -c bioconda sourmash parallel
```
to install the software, and then run
```
conda activate smash
```
to activate the conda environment so you can run the software.
```{note}
Victory conditions: your prompt should start with
`(smash) `
and you should now be able to run `sourmash` and have it output usage information!!
```
## Create a working subdirectory
Make a directory named `kmers` and change into it.
```
mkdir ~/kmers
cd ~/kmers
```
## Download a database and a taxonomy spreadsheet.
We're going to start by doing a reference-based _compositional analysis_ of the lemonade metagenome from [Taylor Reiter's STAMPS 2022 tutorial on assembly and binning](https://github.com/mblstamps/stamps2022/blob/main/assembly_and_binning/tutorial_assembly_and_binning.md).
For this purpose, we're going to need a database of known genomes. We'll use the GTDB genomic representatives database, containing ~65,000 genomes - that's because it's smaller than the full GTDB database (~320,000) or Genbank (~1.3m), and hence faster. But you can download and use those on your own, if you like!
You can find the link to a prepared GTDB RS207 database for k=31 on the [the sourmash prepared databases page](https://sourmash.readthedocs.io/en/latest/databases.html). Let's download it to the current directory:
```
curl -JLO https://osf.io/3a6gn/download
```
This will create a 1.7 GB file:
```
ls -lh gtdb-rs207.genomic-reps.dna.k31.zip
```
and you can examine the contents with sourmash `sig summarize`:
```
sourmash sig summarize gtdb-rs207.genomic-reps.dna.k31.zip
```
which will show you:
```
>path filetype: ZipFileLinearIndex
>location: /home/stamps2022/kmers/gtdb-rs207.genomic-reps.dna.k31.zip
>is database? yes
>has manifest? yes
>num signatures: 65703
>** examining manifest...
>total hashes: 212454591
>summary of sketches:
> 65703 sketches with DNA, k=31, scaled=1000, abund 212454591 total hashes
```
There's a lot of things to digest in this output but the two main ones are:
* there are 65,703 genome sketches in this database, for a k-mer size of 31
* this database represents 212 *billion* k-mers (multiply number of hashes by the scaled number)
If you want to read more about what, exactly, sourmash is doing, please see [Lightweight compositional analysis of metagenomes with FracMinHash and minimum metagenome covers](https://www.biorxiv.org/content/10.1101/2022.01.11.475838v2), Irber et al., 2022.
We also want to download the accompanying taxonomy spreadsheet:
```
curl -JLO https://osf.io/v3zmg/download
```
and uncompress it:
```
gunzip gtdb-rs207.taxonomy.csv.gz
```
This spreadsheet contains information connecting Genbank genome identifiers to the GTDB taxonomy - take a look:
```
head -2 gtdb-rs207.taxonomy.csv
```
will show you:
```
>ident,superkingdom,phylum,class,order,family,genus,species
>GCF_000566285.1,d__Bacteria,p__Proteobacteria,c__Gammaproteobacteria,o__Enterobacterales,f__Enterobacteriaceae,g__Escherichia,s__Escherichia coli
```
Let's index the taxonomy database using SQLite, for faster access later on:
```
sourmash tax prepare -t gtdb-rs207.taxonomy.csv \
-o gtdb-rs207.taxonomy.sqldb -F sql
```
This creates a file `gtdb-rs207.taxonomy.sqldb` that contains all the information in the CSV file, but which is faster to load than the CSV file.
## Download and prepare sample reads
Next, let's download one of the metagenomes from [the assembly and binning tutorial](https://github.com/mblstamps/stamps2022/blob/main/assembly_and_binning/tutorial_assembly_and_binning.md#retrieving-the-data).
We'll use sample SRR8859675 for today, and you can view sample info [here](https://www.ebi.ac.uk/ena/browser/view/SRR8859675?show=reads) on the ENA.
To download the metagenome from the ENA, run:
```
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR885/005/SRR8859675/SRR8859675_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR885/005/SRR8859675/SRR8859675_2.fastq.gz
```
Now we're going to prepare the metagenome for use with sourmash by converting it into a _signature file_ containing _sketches_ of the k-mers in the metagenome. This is the step that "shreds" all of the reads into k-mers of size 31, and then does further data reduction by [sketching](https://en.wikipedia.org/wiki/Streaming_algorithm) the resulting k-mers.
To build a signature file, we run `sourmash sketch dna` like so:
```
sourmash sketch dna -p k=31,abund SRR8859675*.gz \
-o SRR8859675.sig.gz --name SRR8859675
```
Here we're telling sourmash to sketch at k=31, and to track k-mer multiplicity (with 'abund'). We sketch _both_ metagenome files together into a single signature named `SRR8859675` and stored in the file `SRR8859675.sig.gz`.
When we run this, we should see:
>`calculated 1 signature for 3452142 sequences taken from 2 files`
which tells you how many reads there are in these two files!
If you look at the resulting files,
```
ls -lh SRR8859675*
```
you'll see that the signature file is _much_ smaller (2.5mb) than the metagenome files (~600mb). This is because of the way sourmash uses a reduced representation of the data, and it's what makes sourmash fast. Please see the paper above for more info!
Also note that the GTDB prepared database we downloaded above was built using the same `sourmash sketch dna` command, but applied to 65,000 genomes and stored in a zip file.
## Find matching genomes with `sourmash gather`
At last, we have the ingredients we need to analyze the metagenome against GTDB!
* the software is installed
* the GTDB database is downloaded
* the metagenome is downloaded and sketched
Now, we'll run the [sourmash gather](https://sourmash.readthedocs.io/en/latest/command-line.html#sourmash-gather-find-metagenome-members) command to find matching genomes.
Run gather - this will take ~6 minutes:
```
sourmash gather SRR8859675.sig.gz gtdb-rs207.genomic-reps.dna.k31.zip --save-matches matches.zip
```
Here we are saving the matching genome sketches to `matches.zip` so we can rerun the analysis if we like.
The results will look like this:
```
overlap p_query p_match avg_abund
--------- ------- ------- ---------
2.0 Mbp 0.4% 31.8% 1.3 GCF_004138165.1 Candidatus Chloroploc...
1.9 Mbp 0.5% 66.9% 2.1 GCF_900101955.1 Desulfuromonas thioph...
0.6 Mbp 0.3% 23.3% 3.2 GCA_016938795.1 Chromatiaceae bacteri...
0.6 Mbp 0.5% 27.3% 6.6 GCA_016931495.1 Chlorobiaceae bacteri...
...
found 22 matches total;
the recovered matches hit 5.3% of the abundance-weighted query
```
In this output:
* the last column is the name of the matching GTDB genome
* the first column is the estimated overlap between the metagenome and that genome, in base pairs (estimated from shared k-mers)
* the second column, `p_query` is the percentage of metagenome k-mers (weighted by multiplicity) that match to the genome; this will approximate the percentage of _metagenome reads_ that will map to this genome, if you map.
* the third column, `p_match`, is the percentage of the genome k-mers that are matched by the metagenome; this will approximate the percentage of _genome bases_ that will be covered by mapped reads;
* the fourth column is the estimated mean abundance of this genome in the metagenome.
The other interesting number is here:
>`the recovered matches hit 5.3% of the abundance-weighted query`
which tells you that you should expect about 5.3% of the metagenome reads to map to these 22 reference genomes.
```{note}
You can try running gather without abundance weighting:
`sourmash gather SRR8859675.sig.gz matches.zip --ignore-abundance`
How does the output differ?
The main number that changes bigly is:
>`the recovered matches hit 2.4% of the query (unweighted)`
which represents the proportion of _unique_ kmers in the metagenome that are not found in any genome.
This is (approximately) the following number:
* suppose you assembled the entire metagenome perfectly into perfect contigs (**note, this is impossible, although you can get close with "unitigs"**);
* and then matched all the genomes to the contigs;
* approximately 2.4% of the bases in the contigs would have genomes that match to them.
Interestingly, this is the _only_ number in this entire tutorial that is essentially impossible to estimate any way other than with k-mers.
This number is also a big underestimate of the "true" number for the metagenome - we'll explain more later :)
```
## Build a taxonomic summary of the metagenome
We can use these matching genomes to build a taxonomic summary of the metagenome using [sourmash tax metagenome](https://sourmash.readthedocs.io/en/latest/command-line.html#sourmash-tax-subcommands-for-integrating-taxonomic-information-into-gather-results) like so:
```
# rerun gather, save the results to a CSV
sourmash gather SRR8859675.sig.gz matches.zip -o SRR8859675.x.gtdb.csv
# use tax metagenome to classify the metagenome
sourmash tax metagenome -g SRR8859675.x.gtdb.csv \
-t gtdb-rs207.taxonomy.sqldb -F human -r order
```
this shows you the rank, taxonomic lineage, and weighted fraction of the metagenome at the 'order' rank.
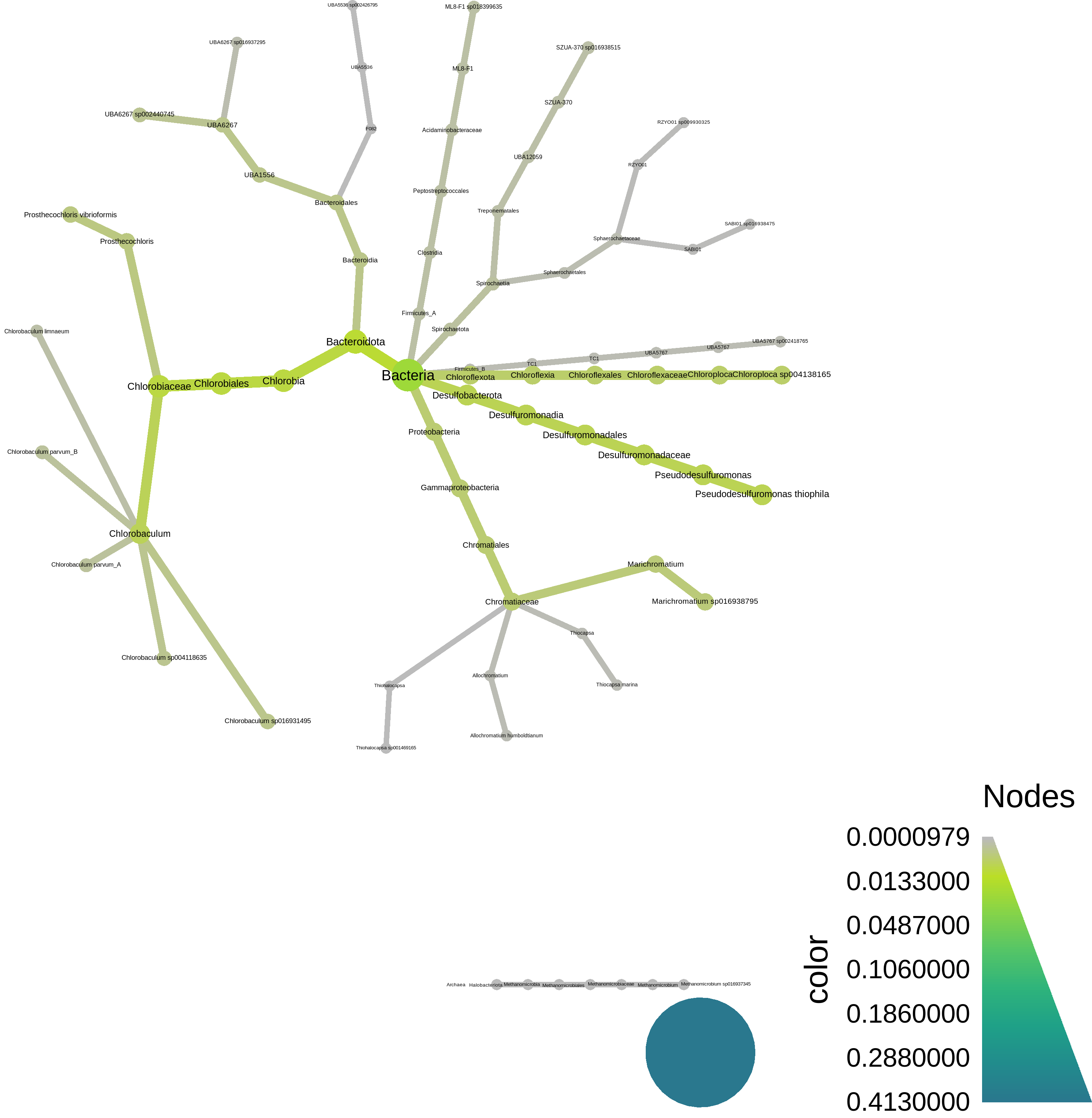
At the bottom, we have a script to plot the resulting taxonomy using [metacoder](https://grunwaldlab.github.io/metacoder_documentation/) - here's what it looks like:
## Interlude: why reference-based analyses are problematic for environmental metagenomes
Reference-based metagenome classification is highly dependent on the organisms present in our reference databases.
For well-studied environments, such as human-associated microbiomes, your classification percentage is likely to be quite high.
In contrast, this is an environmental metagenome, and you can see that we're estimating only 5.3% of it will map to GTDB reference genomes!
Wow, that's **terrible**! Our taxonomic and/or functional analysis will be based on only 1/20th of the data!
What could we do to improve that?? There are two basic options -
(1) Use a more complete reference database, like the entire GTDB, or Genbank. This will only get you so far, unfortunately. (See exercises at end.)
(2) Assemble and bin the metagenome to produce new reference genomes!
There are other things you could think about doing here, too, but these are probably the "easiest" options. And what's super cool is that we did the second one as part of [Taylor Reiter's STAMPS 2022 tutorial on assembly and binning](https://github.com/mblstamps/stamps2022/blob/main/assembly_and_binning/tutorial_assembly_and_binning.md). So can we include that in the analysis??
Yes, yes we can! We can integrate the three MAGs that Taylor generated during her tutorial into the sourmash analysis.
We'll need to:
* download the three genomes;
* sketch them with k=31;
* re-run sourmash gather with both GTDB _and_ the MAGs.
Let's do it!!
## Update gather with information from MAGs
First, download the MAGs:
```
# Download 3 MAGs generated by ATLAS
curl -JLO https://osf.io/fejps/download
curl -JLO https://osf.io/jf65t/download
curl -JLO https://osf.io/2a4nk/download
```
This will produce three files, `MAG*.fasta`.
Now sketch them:
```
sourmash sketch dna MAG*.fasta --name-from-first
```
here, `--name-from-first` is a convenient way to give them distinguishing names based on the name of the first contig in the FASTA file; you can see the names of the signatures by doing:
```
sourmash sig describe MAG1.fasta.sig
```
Now, let's re-do the metagenome classification with the MAGs:
```
sourmash gather SRR8859675.sig.gz MAG*.sig matches.zip -o SRR8859675.x.gtdb+MAGS.csv
```
and look, we classify a lot more!
```
overlap p_query p_match avg_abund
--------- ------- ------- ---------
2.3 Mbp 12.1% 99.9% 39.4 MAG2_1
2.2 Mbp 26.5% 99.9% 92.4 MAG3_1
2.0 Mbp 0.4% 31.8% 1.3 GCF_004138165.1 Candidatus Chloroploc...
1.9 Mbp 0.5% 66.9% 2.1 GCF_900101955.1 Desulfuromonas thioph...
1.0 Mbp 2.7% 100.0% 20.3 MAG1_1
0.6 Mbp 0.3% 23.2% 3.1 GCA_016938795.1 Chromatiaceae bacteri...
0.6 Mbp 0.1% 24.5% 2.1 GCA_016931495.1 Chlorobiaceae bacteri...
...
found 24 matches total;
the recovered matches hit 43.5% of the abundance-weighted query
```
Here we see a few interesting things -
(1) The three MAG matches are all ~100% present in the metagenome.
(2) They are all at high abundance in the metagenome, because assembly needs genomes to be ~5x or more in abundance in order to work!
(3) Because they're at high abundance and 100% present, they account for _a lot_ of the metagenome!
What's the remaining 50%? There are several answers -
(1) most of the constitutent genomes aren't in the reference database;
(2) not everything in the metagenome is high enough coverage to bin into MAGs;
(3) not everything in the metagenome is bacterial or archaeal, and we didn't do viral or eukaryotic binning;
(4) some of what's in the metagenome k-mers may simply be erroneous (although with abundance weighting, this is likely to be a small chunk of things)
## Classify the taxonomy of the MAGs; update metagenome classification
Now we can also classify the genomes and update the taxonomic summary of the metagenome!
First, classify the genomes using GTDB; this will use trace overlaps between contigs in the MAGs and GTDB genomes to tentatively identify the _entire_ bin.
```
for i in MAG*.fasta.sig
do
# get 'MAG' prefix. => NAME
NAME=$(basename $i .fasta.sig)
# search against GTDB
echo sourmash gather $i gtdb-rs207.genomic-reps.dna.k31.zip \
--threshold-bp=5000 \
-o ${NAME}.x.gtdb.csv
done | parallel
```
(This will take about a minute.)
Here, we're using a for loop and [GNU parallel](https://www.gnu.org/software/parallel/) to classify the three genomes in parallel.
If you scan the results quickly, you'll see that one MAG has matches in genus Prosthecochloris, another MAG has matches to Chlorobaculum, and one has matches to Candidatus Moranbacteria.
Let's classify them "officially" using sourmash and an average nucleotide identity threshold of 0.8 -
```
sourmash tax genome -g MAG*.x.gtdb.csv \
-t gtdb-rs207.taxonomy.sqldb -F human \
--ani 0.8
```
This is an extremely liberal ANI threshold, incidentally; in reality you'd probably want to do something more stringent, as at least one of these is probably a new species.
You should see:
```
>sample name proportion lineage
>----------- ---------- -------
>MAG3_1 5.3% d__Bacteria;p__Bacteroidota;c__Chlorobia;o__Chlorobiales;f__Chlorobiaceae;g__Prosthecochloris;s__Prosthecochloris vibrioformis
>MAG2_1 5.0% d__Bacteria;p__Bacteroidota;c__Chlorobia;o__Chlorobiales;f__Chlorobiaceae;g__Chlorobaculum;s__Chlorobaculum parvum_B
>MAG1_1 1.1% d__Bacteria;p__Patescibacteria;c__Paceibacteria;o__Moranbacterales;f__UBA1568;g__JAAXTX01;s__JAAXTX01 sp013334245
```
The proportion here is the fraction of k-mers in the MAG that are annotated.
Now let's turn this into a lineage spreadsheet:
```
sourmash tax genome -g MAG*.x.gtdb.csv \
-t gtdb-rs207.taxonomy.sqldb -F lineage_csv \
--ani 0.8 -o MAGs
```
This will produce a file `MAGs.lineage.csv`; let's take a look:
```
cat MAGs.lineage.csv
```
You should see:
```
>ident,superkingdom,phylum,class,order,family,genus,species
>MAG1_1,d__Bacteria,p__Patescibacteria,c__Paceibacteria,o__Moranbacterales,f__UBA1
568,g__JAAXTX01,s__JAAXTX01 sp013334245
>MAG2_1,d__Bacteria,p__Bacteroidota,c__Chlorobia,o__Chlorobiales,f__Chlorobiaceae,
g__Chlorobaculum,s__Chlorobaculum parvum_B
>MAG3_1,d__Bacteria,p__Bacteroidota,c__Chlorobia,o__Chlorobiales,f__Chlorobiaceae,
g__Prosthecochloris,s__Prosthecochloris vibrioformis
```
And if we re-classify the metagenome using the combined information, we see:
```
sourmash tax metagenome -g SRR8859675.x.gtdb+MAGS.csv \
-t gtdb-rs207.taxonomy.sqldb MAGs.lineage.csv \
-F human -r order
```
Now only 56.5% remains unclassified, which is much better than before!
## Interlude: where we are and what we've done so far
To recap, we've done the following:
* analyzed a metagenome's composition against 65,000 GTDB genomes, using 31-mers;
* found that a disappointingly small fraction of the metagenome can be identified this way.
* incorporated MAGs built from the metagenome into this analysis, bumping up the classification rate to ~45%;
* added taxonomic output to both sets of analyses.
ATLAS only bins bacterial and archaeal genomes, so we wouldn't expect much in the way of viral or eukaryotic genomes to be binned.
But... how much even _assembles_?
Let's pick a few of the matching genomes out from GTDB and evaluate how many of the k-mers from that genome match to the unassembled metagenome, and then how many of them match to the assembled contigs.
First, download the contigs:
```
curl -JLO https://osf.io/jfuhy/download
```
this produces a file `SRR8859675_contigs.fasta`.
Sketch the contigs into a sourmash signature -
```
sourmash sketch dna SRR8859675_contigs.fasta --name-from-first
```
Now, extract one of the top gather matches to use as a query; this is "Chromatiaceae bacterium":
```
sourmash sig cat matches.zip --include GCA_016938795.1 -o GCA_016938795.sig
```
### Evaluate containment of known genomes in reads vs assembly
```{note}
If you want to just start here, you can download the files needed for the below sourmash searches from [this link](https://github.com/mblstamps/stamps2022/raw/main/kmers_and_sourmash/assembly-loss-files.zip).
```
Now do a containment search of this genome against both the unassembled metagenome and the assembled (but unbinned) contigs -
```
sourmash search --containment GCA_016938795.sig \
SRR8859675*.sig* --threshold=0 --ignore-abund
```
We see:
```
similarity match
---------- -----
23.3% SRR8859675
4.7% SRR8859675_0
```
where the first match (at 23.3% containment) is to the metagenome. (You'll note this matches the % in the gather output, too.)
The second match is to the assembled contigs, and it's 4.7%. That means ~19% of the k-mers that match to this GTDB genome are present in the unassembled metagenome, but are lost during the assembly process.
Why?
Some thoughts and answers
It _could_ be that the GTDB genome is full of errors, and those errors are shared with the metagenome, and assembly is squashing those errors. Yay!
But this is extremely unlikely... This GTDB genome was built and validated entirely independently from this sample...
It's much more likely (IMO) that one of two things is happening:
(1) this sample is at low abundance in the metagenome, and assembly can only recover parts of it.
(2) this sample contains _several_ strain variants of this genome, and assembly is squashing the strain variation, because that's what assembly does.
Note, you can try the above with another one of the top gather matches and you'll see it's *entirely* lost in the process of assembly -
```
sourmash sig cat matches.zip --include GCF_004138165.1 -o GCF_004138165.sig
sourmash search --containment GCF_004138165.sig \
SRR8859675*.sig* \
--ignore-abund --threshold=0
```
## Summary and concluding thoughts
Above, we demonstrated a _reference-based_ analysis of shotgun metagenome data using sourmash.
We then _updated our references_ using the MAGs produced from assembly and binning tutorial, which increased our classification rate substantially.
Last but not least, we looked at the loss of k-mer information due to metagenome assembly.
All of these results were based on 31-mer overlap and containment - k-mers FTW!
A few points:
* We would have gotten slightly different results using k=21 or k=51; more of the metagenome would have been classified with k=21, while the classification results would have been more closely specific to genomes with k=51;
* sourmash is a nice one-stop-shop tool for doing this, but you could have gotten similar results by using other tools.
* Next steps here could include mapping reads to the genomes we found, and/or doing functional analysis on the matching genes and genomes.
|
